RapidShapes computes the shapes above a specified alpha-threshold
by generating
a list of promising
shapes and constructing specialized folding programs for each shape to compute its share of Boltzmann probability.
This aims at a heuristic improvement of runtime, while still computing exact probability values.
Because of many independent components of RapidShapes, the runtime can be decrease
once more, during parallel computation. Thus, probabilistic shape analysis becomes feasible in medium-scale applications,
such as the screening of RNA transcripts in a bacterial genome.
The RapidShape program uses a pipeline to predict exact probabilities of RNA shapes.
This pipeline contains several steps:
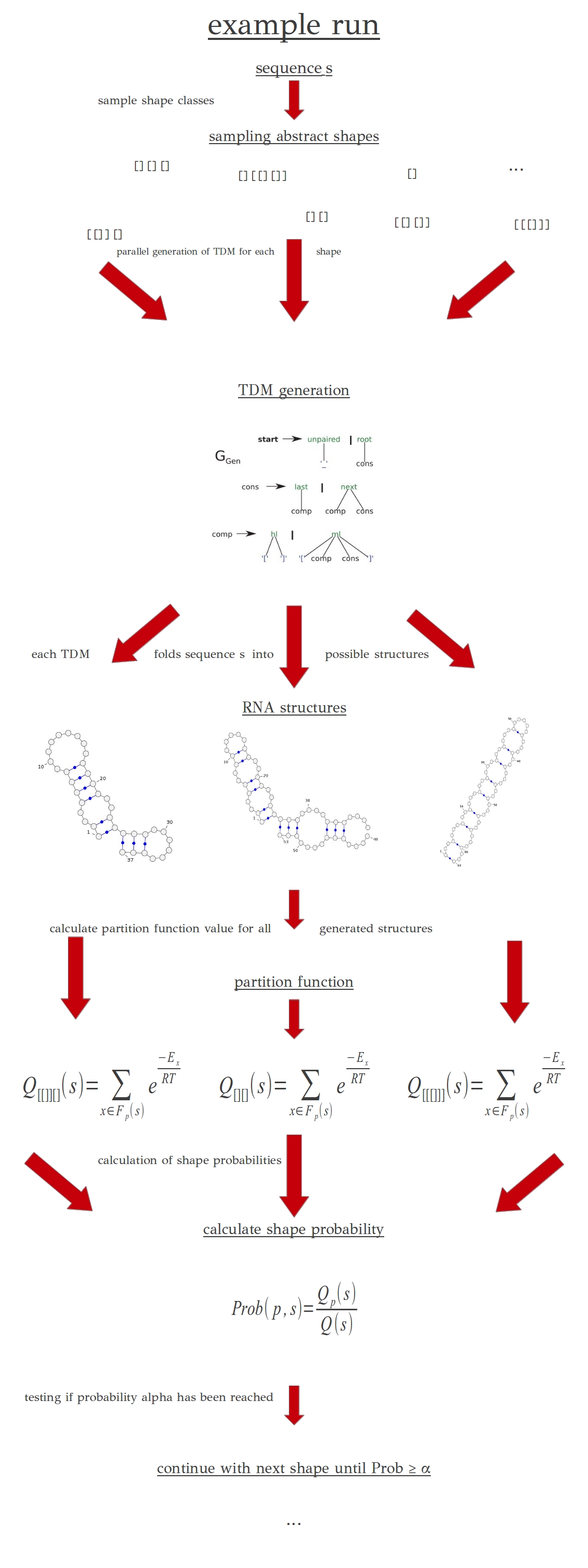 Figure 1: Illustration gives an overview of probability calculation for a shape.
Q
p(s) is the sum of partition function values for a specific shape class with the parameters:
s - sequence
Figure 1: Illustration gives an overview of probability calculation for a shape.
Q
p(s) is the sum of partition function values for a specific shape class with the parameters:
s - sequence
E
x - energy of structure x in kcal/mol
R - universal gas constant (0.00198717 kcal/K)
T - temperature in Kelvin
F
p(s) - a set of all possible shapes the sequence s could be fold in by specific TDM
Prob(p,s) is the probability of a specific shape class Q
p(s) with the parameters:
Q
p(s) - partition function value of a specific shape class
Q(s) - partition function value of the complete folding space
Note: this figure is simplified for better understanding.
At first rapidshapes creates a list of sampled abstract RNA shapes. Abstract shape classes are used, because many different concrete RNA shapes can be described as one abstract shape. The advantage of that is used in the next step.
For each abstract shape class there will be generate a specific thermodynamic matcher. For that a tree grammar
is used to describe the structures.
Tree grammars make explicit the semantics of each grammar rule, and can be compiled directly into executable
code using the algebra dynamic programming technology (ADP).
This specific ADP grammar becomes translated into C++ code during the GAPc compiler.
Supported by this method shape specific TDM's are generated.
In the next step the C++ code becomes compiled into an executable binary by the gcc compiler.
These shape specific TDM's fold the given sequence
s into all possible structures which fit into the abstract shape class the TDM were made from.
Every structure gets ranked by the partition function and the values of all structures of a specific shape get accumulated to calculate the probability of the whole shape class.
Thus, rapid shapes computes the probability of a sub-foldingspace by division of the partition function value of an sub-foldingspace by the partition function value of the complete folding space.
While the specified probability
alpha is not reached yet, the program continues calculating probabilities of sub-foldingspaces.
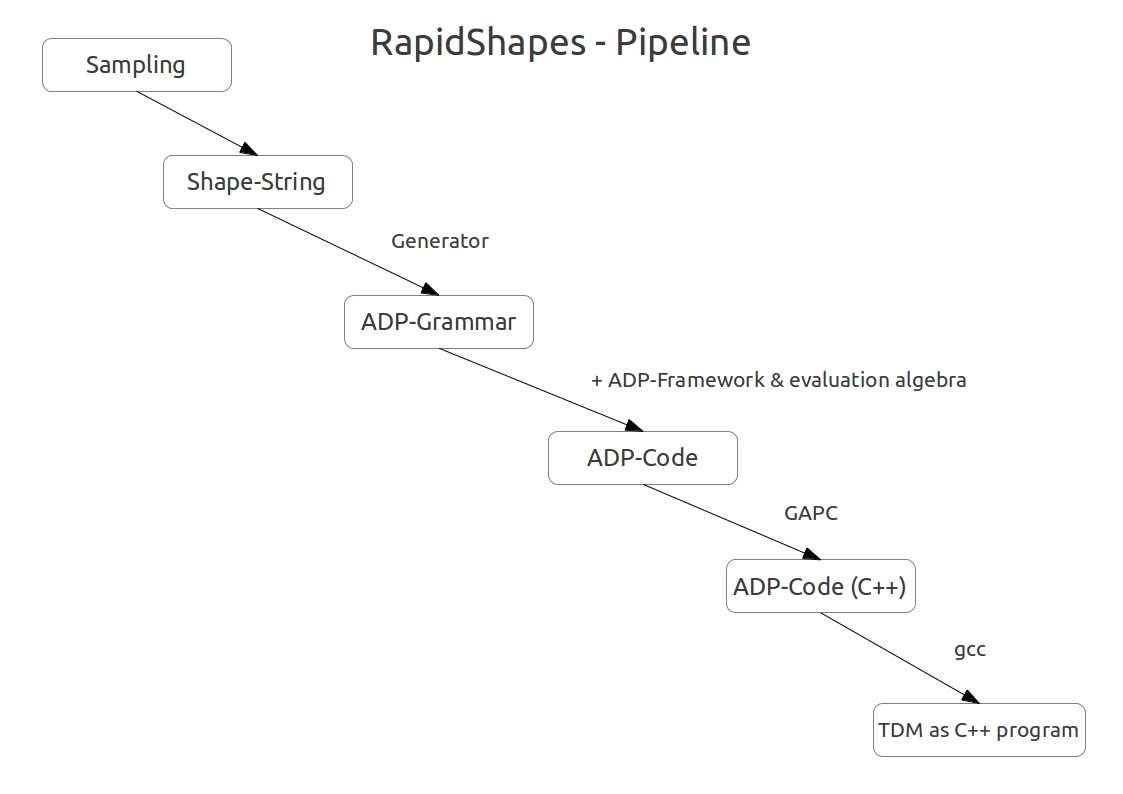
Figure 2: Shows the exact steps form shape-sampling to TDM generation.
The first step, the sampling, can be replaced by other methods to gain a initial set of promising shape classes whose proability is afterwards exactly computed by the rest of the program. The following four different functions allow for alternative shape guessing methods (sample, kbest, subopt and list):
Estimate shape frequencies via sampling a specific number of secondary structure from the folding-space, via stochastical backtracing.
A simple shape class analysis is performed and the k-best
energetically ordered shape classes are selected.
similar to "kbest". Instead of the k energetically best shape
classes, those shape classes are used whoes energy deviates up to a
certain threshlold from minimal free energy for the input sequence.
If you have an alternative method of guessing shapes, you can also
provide a list of these shape classes. Take care, that your input
sequence can fold into these shapes at all!
| alpha |
RapidShapes computes individual shape class probabilities until either alpha percent of the folding space is explored or no more guessed shape classes are uncomputed. We suggest an alpha of 90% or less.
|
| number of samples |
Sets the number of samples that are drawn to estimate shape probabilities.
In our experience, 1000 iterations are sufficient to achieve reasonable results for shapes with high probability. Thus, default is 1000.
|
| kbest |
RapidShapes will first perform a simple shape analysis for the best 'kbest' shapes. Choice of an appropriate value for kbest is not easy, since it depends on sequence length and base composition.
|
| relative deviation |
relative deviation sets the energy range as percentage value of the minimum free energy. For example, when relative deviation is specified as 5.0, and the minimum free energy is -10.0 kcal/mol, the energy range is set to -9.5 to -10.0 kcal/mol.
relative deviation must be a positive floating point number; by default it is set to to 10 %.
It cannot be combined with absolute deviation.
|
| absolute deviation |
This sets the energy range as an absolute value of the minimum free energy. For example, when absolute deviation 10.0 kcal/mol is specified, and the minimum free energy is -10.0 kcal/mol, the energy range is set to 0.0 to -10.0 kcal/mol.
absolute deviation must be a positive floating point number. Cannot be combined with relative deviation.
|
| list |
You might want to manually provide a list of shape classes that should be checked via TDMs. Individual shapes are separated by whitespaces, commas or semicolons.
An example input might be "[],[[][]],[][]"
|
| grammar |
How to treat dangling end energies for bases adjacent to helices in free ends and multi-loops.
nodangle: (-d 0 in Vienna package) ignores dangling energies altogether.
overdangle: (-d 2 in Vienna package) always dangles bases onto helices, even if they are part of neighbouring helices themselves. Seems to be wrong, but could perform surprisingly well.
microstate: (-d 1 in Vienna package) correct optimization of all dangling possibilities, unfortunately this results in an semantically ambiguous search space regarding Vienna-Dot-Bracket notations.
macrostate: (no correspondens in Vienna package) same as microstate, while staying unambiguous. Unfortunately, mfe computation violates Bellman's principle of optimality.
See [jan:schud:ste:gie:2011] for further details.
|
| shape level |
shape level is the level of abstraction or dissimilarity which defines a different shape. In general, helical regions are depicted by a pair of opening and closing brackets and unpaired regions are represented as a single underscore. The differences of the shape types are due to whether a structural element (bulge loop, internal loop, multiloop, hairpin loop, stacking region and external loop) contributes to the shape representation: Five types are implemented. Their differences are shown in the following example:
CGUCUUAAACUCAUCACCGUGUGGAGCUGCGACCCUUCCCUAGAUUCGAAGACGAG
((((((...(((..(((...))))))...(((..((.....))..)))))))))..
| 1 |
Most accurate - all loops and all unpaired |
[_[_[]]_[_[]_]]_ |
| 2 |
Nesting pattern for all loop types and unpaired regions in external loop and multiloop |
[[_[]][_[]_]] |
| 3 |
Nesting pattern for all loop types but no unpaired regions |
[[[]][[]]] |
| 4 |
Helix nesting pattern in external loop and multiloop |
[[][[]]] |
| 5 |
Most abstract - helix nesting pattern and no unpaired regions |
[[][]] |
The following image also describes the differences between shape types:
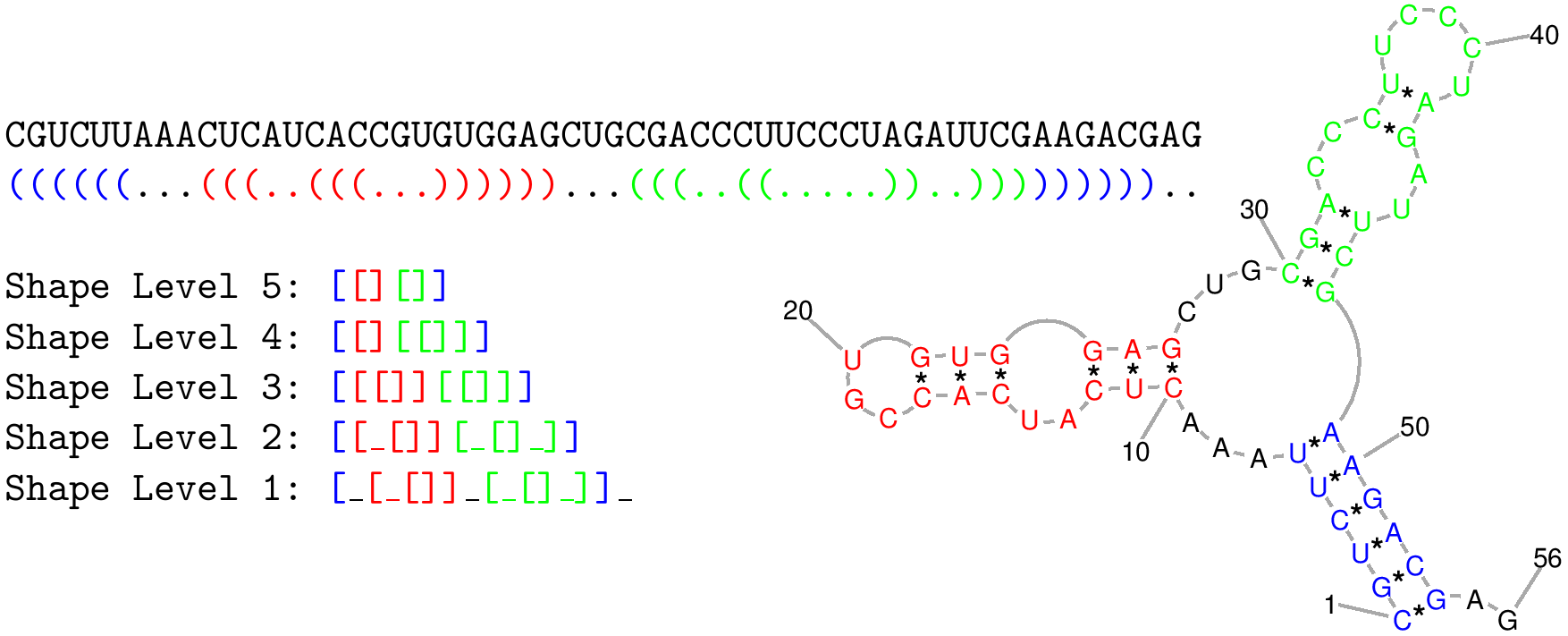
|
| temperature |
The energy parameters used in the calculation have been measured at 37 C. Parameters at other temperatures can be extrapolated, but for temperatures far from 37 C results will be increasingly unreliable.
|
| thermodynamic model parameters |
Read energy parameters from a file, instead of using the default parameter set. See the RNAlib (Vienna RNA package) documentation for details on the file format.
Default are parameters released by the Turner group in 2004 (see [mat:dis:chil:schro:zuk:tur:2004] and [tur:mat:2010]). A visit of the aforementioned author's Nearest Neighbor Database might also be informative.
|
| lonely base pairs |
Lonely base pairs have no stabilising effect, because they cannot stack on another pair, but they heavily increase the size of the folding space. Thus, we normally forbid them. Should you want to allow them set lonely base pairs to 1.
lonely base pairs must be either 0 (=don't allow lonely base pairs) or 1 (= allow them).
Default is 0, i.e. no lonely base pairs.
|
| decimals for probabilities |
Sets the number of digits used for printing shape probabilities.
decimals for probabilities must be a positive integer number. The default value is 7.
|
