|
Attention:
Due to technical maintenance some tools might be unavailable. See maintenance information. 
|

|

|
|
|








|





















ChromA - Manual
Introduction
ChromA performs a retention time alignment of two chromatograms of mass spectra, as produced by GC/LC-MS experiments. The alignment is calculated by the commonly known algorithm Dynamic Time Warping (DTW), which is a continuous generalization of classical alignment of discrete sequences, such as strings. It runs in quadratic space and time, depending on the size of the input data, in this case the number of scans of a chromatogram. DTW is used to align mass spectra of two GC/LC-MS chromatograms to the time domain of the first chromatogram. Additional parameters, such as different local similarity/distance functions on mass spectral intensities, positions of known compounds (anchors) and windows around these known compounds (neighborhood) allow good or even optimal alignments concerning the global minimization of a local distance or global maximization of a local similarity function to be found in considerably less time.
Chromatograms
Currently supported file formats are AIA/ANDIMS netcdf with filename suffixes .cdf or .nc and mzXML with file suffix .mzxml. Other file suffixes will not work! ChromA should also work on processed chromatograms, which have been baseline corrected and deconvoluted. You can try to use the alignment on peak extracted chromatograms, but be aware that Dynamic Time Warping expects continuous data.
File Formats
Required variables/fields to be contained in the netcdf files are:
- total_intensity
- mass_values
- intensity_values
- scan_index
- scan_acquisition_time
- mass_range_min
- mass_range_max
Please note, that ChromA will not work from the web frontend with a different naming scheme!
Anchors
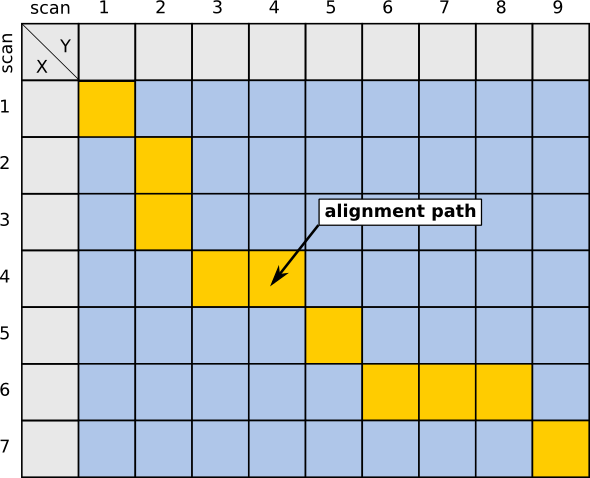
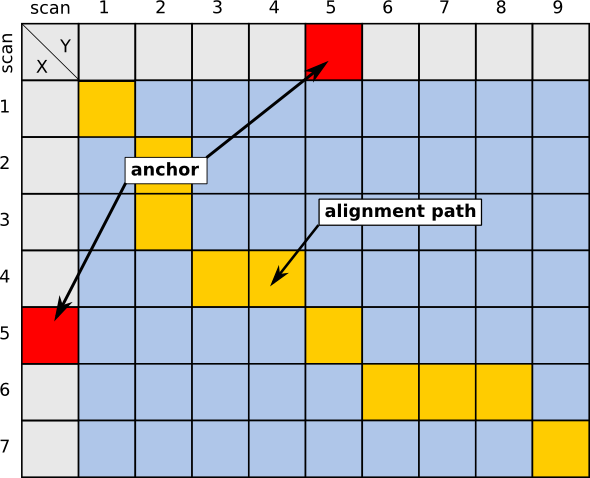
In the context of Chromatography, anchors can be identified substances, e.g. in GC, homologue alkanes, used to calculate retention indices, as well as MS/MS identifications in LC. This additional information can then be used to constrain the area of the alignment, thereby speeding up the calculation. The images below give a visual imemssion of this speedup.
 |
 |
 |
| Unconstrained case | Using 1 anchor | Calculations only in colored rectangular bounds |
Format for Anchor Input
Anchors are entered into the web form in a space separated
format, giving the scan number for each anchor in increasing
order. E.g.: 40 84 163 231
The number of anchors for both chromatograms must be the same to
ensure a one-to-one matching.
Format for file based Anchor Input (WebStart)
The file format follows basic conventions for tab separated value data. The first line starts with a > character, immediately followed by the filename of the corresponding chromatogram file. The name can be prefixed by an absolute path, e.g. on Windows starting with C:\ or / on unix like systems. The next line holds the column names, each name separated by a tab from its next neighbor to the right.
Current code of conduct is to name the first column Name, the second one RI for retention index information, the third one RT for retention time and the last one Scan for the scan index of the apex of the anchor.
The name of an anchor must be unique among all defined anchor files, so that e.g. Anchor1 in anchorsExp1.txt is supposed to be identical to Anchor1 in anchorsExp2.txt and so on.
The only mandatory columns are Name and Scan, missing values can be indicated by - (minus).
Preprocessing
The only emprocessing done by ChromA is the binning of m/z values, such that an even grid is produced and m/z bins can be directly indexed by integers. Each m/z channel (e.g. 55.0 - 55.99) is currently resolved with a width of one (our example as index 55). Multiple intensities falling into the same bin are added. This allows immediate use of the intensity profile vector of each mass spectrum for the local similarity/distance calculations.
You can exclude all intensities contained in a mass channel throughout the whole chromatogram by entering the mass indices to be excluded in the web form under Mass Filter.
Format for Mass Filter Input
Mass Filter Input is a space separated list of m/z-bins which you would like to exclude from the alignment. E.g.: 70 71 72 73 would remove all signals within mass bins greater or equal to 70 and smaller than 74.
Dynamic Time Warp Parameters
Local distance/similarity
The local distance or similarity function which is calculated between intensity profile vectors.
The ChromA web interface provides different local functions:
- Euclidean distance
- Cosine similarity
- Linear correlation
- Hamming distance
- TIC squared distance
We generally recommend using either cosine similarity or linear correlation, these tend to produce the most meaningful alignments. Euclidean distance produces rather smooth warpings, a fast, but less exact alternative is Hamming distance. TIC squared distance is much quicker to compute than the other ones, but it should only be used for quick evaluation and is only included for completeness.
Neighborhood Radius
The integer neighborhood radius around each anchor, which should be considered by the alignment algorithm. E.g.: 10 means: Consider 10 scans to the left of each anchor, 10 to the right, 10 above and 10 below, giving an area of size ((2x10)+1)2 around each anchor.
 |
| Example, using a neighborhood radius of 1 around anchor. |
Maximum allowed percentage of scan lag
This constraint allows to define a maximum allowed lag in scans between two chromatograms, possibly further limiting the number of pairwise evaluations to be computed by ChromA. It is given as a fractional percentage, so 0.1 would run ChromA with a maximum of 500 scans absolute difference to consider, maximum deviation of 250, both to the left and to the right of the diagonal, if both chromatograms have 5000 scans.
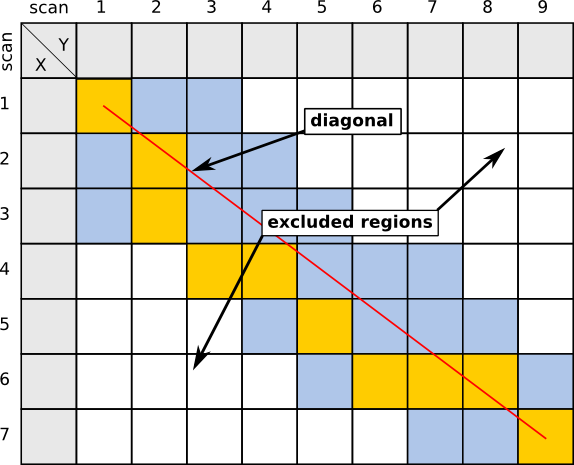 |
| Example, using a percentage of 0.5 as allowed lag, resulting in about five scans allowed lag between chromatograms. |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |

|
|

|